1-1. Prepare Training Data¶
In this notebook, we go through steps to :
embed the population size measurement into the
AnnData.uns['pop'].rename timepoint obs key
perform dimension reduction : diffusion map
sample local state transition
train test split
%load_ext autoreload
%autoreload 2
import os, sys
if sys.platform.startswith("darwin"):
os.environ['KMP_DUPLICATE_LIB_OK']='True'
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import scanpy as sc
sc.settings.set_figure_params(frameon=False, dpi=30)
import our python package as pdp (pseudo dynamics plus)
import pseudodynamics as pdp
os.chdir(pdp.main_dir)
print("workding directory changed to:", pdp.main_dir)
workding directory changed to: /Users/weizhongzheng/Documents/python_project/pseudodynamics_plus
mouse in vivo BM haematopoiesis¶
In this tutorial, we use a special persistent labling single-cell data from Kucinski & Barile et. al, Cell Stem Cell. 2024.
Persistent labeling technique can induces fluorescence in Hoxb5-expressing cells and propogate the label to their progenies. As a result, we can identify offsprings of haematopoitic stem and progenitor cells (HSPC) produced after induction from the by checking the tomato fluorescence (Tom+).
This dataset contains 130700 cells from 9 timepoints spanning 9 month’s time. At each time point we counted the total amount of Tom+ cells in the bone marrow of the mice.
The data is available at figshare 👈, or you can download it with the bash script data_downloading.sh.
# we can download the expression matrix together with experimental settings
# create a data folder
if not os.path.exists("data"):
os.mkdir("data")
adata_full = sc.read_h5ad("data/tompos/combined_filt.h5ad")
adata_full
AnnData object with n_obs × n_vars = 130700 × 4814
obs: 'n_genes', 'n_counts', 'mt_count', 'mt_frac', 'doublet_scores', 'predicted_doublets', 'xist_logn', 'Ygene_logn', 'xist_bin', 'Ygene_bin', 'sex_adata', 'biosample_id', 'cellid', 'RBG', 'SLXid', 'index', '10xsample_description', 'sex_mixed', 'sex_meta', 'mouse_id', 'sortedcells', 'expected_cells_10x', 'cellranger_cellsfound', 'chemistry', 'tom', 'expdate', 'batch', 'timepoint_tx_days', 'start_age', 'sample_id', 'countfile', 'S_score', 'G2M_score', 'phase', 'leiden', 'SLX', 'plate_sorted', 'plate_rearranged', 'well_sorted', 'well_rearranged', 'set_index', 'CI_index', 'mouse_platelabel', 'sort_method', 'sample.name', 'population', 'sex', 'countfolder', 'batch_plate_sorted', 'data_type', 'sex_combined', 'longname', 'anno_man', 'leiden_DM', 'HSCscore', 'nn_HSCscore', 'isroot', 'dpt_pseudotime'
var: 'symbol', 'gene_ids-10x', 'feature_types-10x', 'genome-10x', 'n_counts-10x', 'highly_variable-10x', 'means-10x', 'dispersions-10x', 'dispersions_norm-10x', 'gene_removed-10x', 'mean-10x', 'std-10x', 'geneid-SS2', 'chromosome-SS2', 'is_mito-SS2', 'highly_variable', 'mean', 'std'
uns: 'DM', 'anno_man_colors', 'biosample_id_colors', 'data_type_colors', 'diffmap_evals', 'iroot', 'isroot_colors', 'leiden', 'leiden_DM_colors', 'leiden_DM_sizes', 'leiden_colors', 'leiden_sizes', 'neighbors', 'paga', 'pagaDM', 'pagaPCA', 'pca', 'phase_colors', 'predicted_doublets_colors', 'rank_genes_groups', 'umap_init_pos', 'umap_init_pos_DM'
obsm: 'X_diffmap', 'X_pca', 'X_pca_harmony', 'X_umap', 'X_umap_2d', 'X_umap_3d', 'X_umap_DM_2d', 'X_umap_DM_3d'
varm: 'PCs'
obsp: 'DM_connectivities', 'DM_distances', 'connectivities', 'distances'
Data overview and filtering¶
The preprocess single-cell landscape is batch-correted and cell type annotated.
sc.pl.umap(adata_full,
color=['timepoint_tx_days', 'tom', 'dpt_pseudotime',
'batch', 'anno_man', 'biosample_id', ],
ncols=3, wspace=0.3)
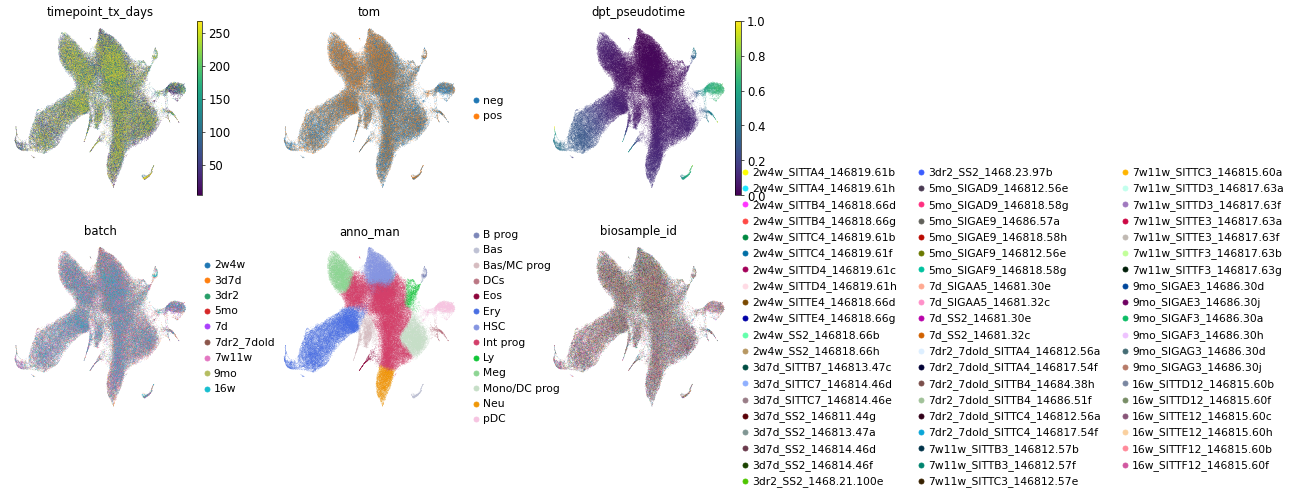
filtering Tom+ cells
For this particular data, the newly generated cells are labeled with fluorensence. We thus filtered Tomato-positive (Tom+) cells and count the number of all the Tom+ cells in the bone marrow. At each time point, we sequenced and FACS-counted cells from several mouse.
# select positive cells and saved under data
adata = adata_full[adata_full.obs['tom'] == 'pos'].copy()
adata.write_h5ad('data/tom_pos.h5ad')
Important !!!
To train the model, you need to placed the processed data under data.
1. Attaching population size measurement¶
One of the main innovations of pseudodyanmics+ is the use of population/tissue size measurement that comes from additional experiment, considering single-cell experiment has the issue of limiting capacity and sampling bias.
However, for dataset that without tissue size measurement, we can go back to sequenced cell number in the sc library. The parameters inferred in this manner may fail to reflect the absolute tissue growth and flux transiting between cell state.
The population info is stored as a dictionary and saved in adata.uns['pop']. The pop dictionary must contain the following keys:
't': the real experiment time point when the cells are counted and sequenced (assume on the same day)'mean': the mean cell number of different experimental repeats for each time point'std': the standard derivation of different experimental repeats'n_lib': the number of repeats
For example, a single cell data with 3 time points (say day0, day4, day10) and 3 repeats each time looks like this:
adata.uns['pop'] = {
"t" = np.array([0, 4, 10]),
"mean" = np.array([mean_d0, mean_d4, mean_d10]),
"var" = np.array([var_d0, var_d4, var_d10]),
"std" = np.array([std_d0, std_d4, std_d10]),
"n_lib" = np.array([3, 3, 3])
}
adata = sc.read_h5ad('data/tom_pos.h5ad')
adata
AnnData object with n_obs × n_vars = 49390 × 4814
obs: 'n_genes', 'n_counts', 'mt_count', 'mt_frac', 'doublet_scores', 'predicted_doublets', 'xist_logn', 'Ygene_logn', 'xist_bin', 'Ygene_bin', 'sex_adata', 'biosample_id', 'cellid', 'RBG', 'SLXid', 'index', '10xsample_description', 'sex_mixed', 'sex_meta', 'mouse_id', 'sortedcells', 'expected_cells_10x', 'cellranger_cellsfound', 'chemistry', 'tom', 'expdate', 'batch', 'timepoint_tx_days', 'start_age', 'sample_id', 'countfile', 'S_score', 'G2M_score', 'phase', 'leiden', 'SLX', 'plate_sorted', 'plate_rearranged', 'well_sorted', 'well_rearranged', 'set_index', 'CI_index', 'mouse_platelabel', 'sort_method', 'sample.name', 'population', 'sex', 'countfolder', 'batch_plate_sorted', 'data_type', 'sex_combined', 'longname', 'anno_man', 'leiden_DM', 'HSCscore', 'nn_HSCscore', 'isroot', 'dpt_pseudotime', 'leiden_orig', 'logk', 'net_prolif', 'log10SR', 'log_density_at_E3', 'log_density_at_E7', 'log_density_at_E12', 'log_density_at_E12_clip', 'log_density_at_E3_clip', 'log_density_at_E7_clip', 'log_density_at_E27', 'log_density_at_E27_clip', 'log_density_at_E49', 'log_density_at_E49_clip', 'log_density_at_E76', 'log_density_at_E76_clip', 'log_density_at_E112', 'log_density_at_E112_clip', 'log_density_at_E161', 'log_density_at_E161_clip', 'log_density_at_E269', 'log_density_at_E269_clip', 'pseudo_bulk', 'rs50', 'split', 'fine_anno'
var: 'symbol', 'gene_ids-10x', 'feature_types-10x', 'genome-10x', 'n_counts-10x', 'highly_variable-10x', 'means-10x', 'dispersions-10x', 'dispersions_norm-10x', 'gene_removed-10x', 'mean-10x', 'std-10x', 'geneid-SS2', 'chromosome-SS2', 'is_mito-SS2', 'highly_variable', 'mean', 'std'
uns: 'DM', 'DM_EigenValues', 'anno_man_colors', 'biosample_id_colors', 'data_type_colors', 'density_predictor', 'diffmap_evals', 'fine_anno_colors', 'iroot', 'isroot_colors', 'leiden', 'leiden_DM_colors', 'leiden_DM_sizes', 'leiden_colors', 'leiden_sizes', 'neighbors', 'original_pop', 'paga', 'pagaDM', 'pagaPCA', 'pca', 'phase_colors', 'pop', 'predicted_doublets_colors', 'pseudo_bulk', 'pseudo_bulk_settings', 'rank_genes_groups', 'rs50', 'umap_init_pos', 'umap_init_pos_DM'
obsm: 'DM_EigenVectors', 'DM_scaled', 'Delta_DM', 'TIGON_u', 'X_diffmap', 'X_pca', 'X_pca_harmony', 'X_pca_scaled', 'X_umap', 'X_umap_2d', 'X_umap_3d', 'X_umap_DM_2d', 'X_umap_DM_3d', 'mellon_u', 'surrogate_u'
varm: 'PCs'
obsp: 'DM_Kernel', 'DM_Similarity', 'DM_connectivities', 'DM_distances', 'connectivities', 'distances'
load FACS data¶
Here we are loading the FACS experiment data. The BM cells of the same mouse are sorted for tomato positive and count total cell number before sending for sequencing.
pop_df = pd.read_csv("data/tompos/model_input_tompos.csv")
pop_df.head()
| biosample_id | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ... | 20 | 24 | 25 | 26 | 28 | sc_ncells_filt | flow_total | start_age | time | sc_ncells_total | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 2w4w_SITTC4_146819.61b | 42 | 16 | 8 | 5 | 15 | 7 | 3 | 19 | 28 | ... | 0 | 0 | 0 | 0 | 0 | 157 | 634.907992 | young | 27 | 161 |
| 1 | 2w4w_SITTC4_146819.61f | 184 | 122 | 103 | 109 | 118 | 97 | 73 | 149 | 146 | ... | 4 | 11 | 2 | 5 | 2 | 1304 | 2320.519107 | young | 27 | 1357 |
| 2 | 2w4w_SITTD4_146819.61c | 257 | 122 | 107 | 76 | 116 | 120 | 78 | 175 | 161 | ... | 7 | 4 | 3 | 2 | 1 | 1422 | 3535.791587 | young | 27 | 1449 |
| 3 | 2w4w_SITTD4_146819.61h | 98 | 61 | 42 | 45 | 42 | 26 | 28 | 52 | 76 | ... | 2 | 2 | 0 | 1 | 0 | 547 | 895.421264 | young | 27 | 563 |
| 4 | 2w4w_SITTE4_146818.66d | 35 | 10 | 10 | 9 | 6 | 16 | 4 | 19 | 16 | ... | 0 | 0 | 0 | 1 | 0 | 137 | 456.845376 | young | 12 | 143 |
5 rows × 26 columns
We summarize the total cell number over different mouse for each time point
pop_by_time = pop_df.groupby("time").agg({"flow_total":["mean", "std","count"], "sc_ncells_filt":["mean", "std"]})
pop_by_time
| flow_total | sc_ncells_filt | ||||
|---|---|---|---|---|---|
| mean | std | count | mean | std | |
| time | |||||
| 3 | 147.000000 | 45.745674 | 4 | 101.00 | 36.487441 |
| 7 | 388.645708 | 254.329995 | 6 | 150.50 | 69.856281 |
| 12 | 576.692900 | 169.724308 | 4 | 155.25 | 106.859331 |
| 27 | 1846.659988 | 1347.949806 | 4 | 857.50 | 606.941238 |
| 49 | 3123.277705 | 2110.007646 | 4 | 1143.00 | 813.058833 |
| 76 | 8233.932637 | 446.605979 | 2 | 2678.00 | 560.028571 |
| 112 | 13949.548025 | 6560.283561 | 4 | 3003.25 | 1733.310392 |
| 161 | 4941.850616 | 3795.978265 | 4 | 1854.25 | 1635.804262 |
| 269 | 41722.985970 | 1788.570460 | 2 | 4320.75 | 1446.069241 |
import matplotlib.pyplot as plt
def plot_mean_std(col_name, ax):
time_points = pop_by_time.index
mean_values = pop_by_time[(col_name, 'mean')]
std_values = pop_by_time[(col_name, 'std')]
ax.plot(range(len(time_points)), mean_values, marker='o', label=col_name)
ax.fill_between(range(len(time_points)),
mean_values - std_values,
mean_values + std_values,
alpha=0.3, color='gray', label=col_name+"_error")
ax.set_xticks(range(len(time_points)))
ax.set_xticklabels(time_points)
fig = plt.figure(dpi=80, figsize=(6, 3))
ax = fig.gca()
plot_mean_std('flow_total', ax=ax)
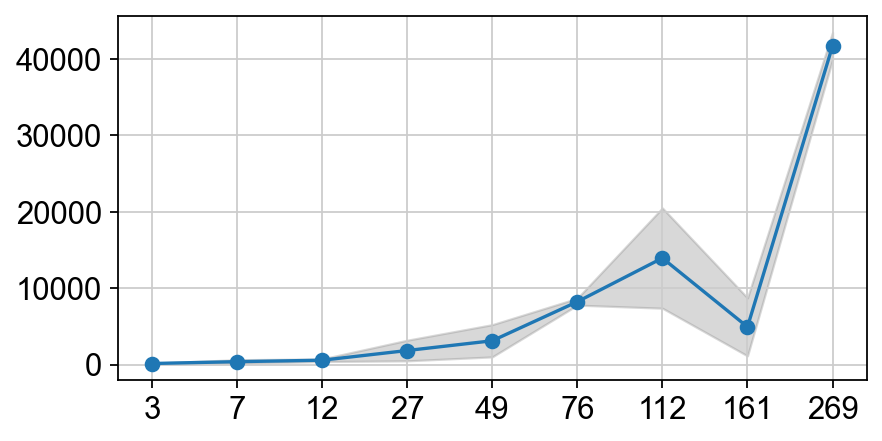
Here we found that the measurment at Day 161 seems to be an outlier. We do the following correction :
we estimate the average growth rate from Day112 to Day269 with \( N_t = N_0 * e^{g \Delta t}\)
next we impute the mean pop size at Day161 with the average growth rate
time_points = pop_by_time.index
mean_values = pop_by_time[('flow_total', 'mean')].values
g = np.log(mean_values[-1] / mean_values[-3]) / (time_points[-1] - time_points[-3])
print(f'the estimated mean g is {g:.5f}')
impute_Day161 = np.exp(g*(time_points[-2] - time_points[-3]) ) * mean_values[-3]
print(f'the imputed mean pop size is {impute_Day161:.2f}')
# replace the mean value
mean_values[-2] = impute_Day161
the estimated mean g is 0.00698
the imputed mean pop size is 19636.45
fig = plt.figure(dpi=80, figsize=(6, 3))
ax = fig.gca()
plot_mean_std('flow_total', ax)
plot_mean_std('sc_ncells_filt', ax)
ax.legend()
<matplotlib.legend.Legend at 0x3264a7f50>
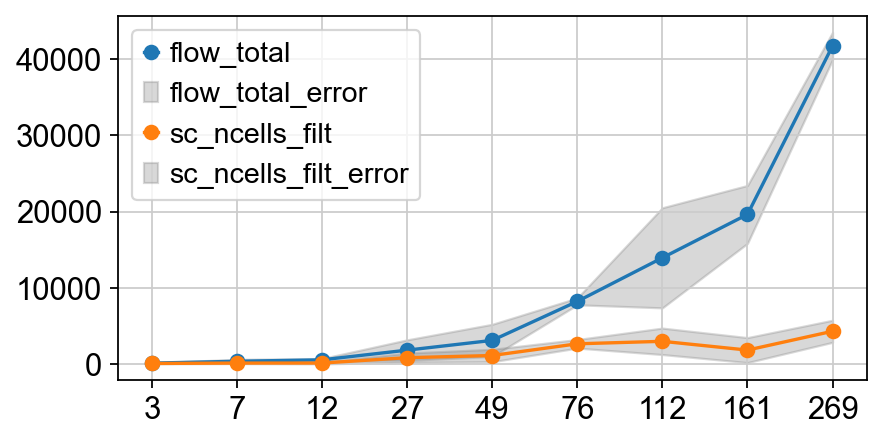
# assign to uns['pop']
adata.uns['pop'] = {
't' : time_points.to_numpy(),
'mean' : mean_values,
'var' : pop_by_time[('flow_total', 'std')].values ** 2, # variance per timepoint
'std' : pop_by_time[('flow_total', 'std')].values,
'n_lib' : pop_by_time[('flow_total', 'count')].values
}
adata.uns['pop']
2. rename Timepoint Key¶
We use a unique obs key timepoint_tx_days to store the timepoint.
if 'timepoint_tx_days' not in adata.obs_keys():
adata.obs['timepoint_tx_days'] = adata.obs['day'].astype(int) # example
check if the time point from the pop info match with that in the sequencing data
adata.obs['timepoint_tx_days'].isin(adata.uns['pop']['t']).all()
True
3. Compute diffusion map (DM)¶
The example dataset has pre-computed diffusion maps. For reproducibility, plesae keep the original diffusion map and pseudotime.
Here, for demonstration, we show two popular methods of getting DM cooridnates and pseudotime.
scanpy diffusion map and dpt_psuedotime
palantir diffusion map and palantir pseudotime
adata
AnnData object with n_obs × n_vars = 49390 × 4814
obs: 'n_genes', 'n_counts', 'mt_count', 'mt_frac', 'doublet_scores', 'predicted_doublets', 'xist_logn', 'Ygene_logn', 'xist_bin', 'Ygene_bin', 'sex_adata', 'biosample_id', 'cellid', 'RBG', 'SLXid', 'index', '10xsample_description', 'sex_mixed', 'sex_meta', 'mouse_id', 'sortedcells', 'expected_cells_10x', 'cellranger_cellsfound', 'chemistry', 'tom', 'expdate', 'batch', 'timepoint_tx_days', 'start_age', 'sample_id', 'countfile', 'S_score', 'G2M_score', 'phase', 'leiden', 'SLX', 'plate_sorted', 'plate_rearranged', 'well_sorted', 'well_rearranged', 'set_index', 'CI_index', 'mouse_platelabel', 'sort_method', 'sample.name', 'population', 'sex', 'countfolder', 'batch_plate_sorted', 'data_type', 'sex_combined', 'longname', 'anno_man', 'leiden_DM', 'HSCscore', 'nn_HSCscore', 'isroot', 'dpt_pseudotime', 'leiden_orig', 'logk', 'net_prolif', 'log10SR', 'log_density_at_E3', 'log_density_at_E7', 'log_density_at_E12', 'log_density_at_E12_clip', 'log_density_at_E3_clip', 'log_density_at_E7_clip', 'log_density_at_E27', 'log_density_at_E27_clip', 'log_density_at_E49', 'log_density_at_E49_clip', 'log_density_at_E76', 'log_density_at_E76_clip', 'log_density_at_E112', 'log_density_at_E112_clip', 'log_density_at_E161', 'log_density_at_E161_clip', 'log_density_at_E269', 'log_density_at_E269_clip', 'pseudo_bulk', 'rs50', 'split', 'fine_anno'
var: 'symbol', 'gene_ids-10x', 'feature_types-10x', 'genome-10x', 'n_counts-10x', 'highly_variable-10x', 'means-10x', 'dispersions-10x', 'dispersions_norm-10x', 'gene_removed-10x', 'mean-10x', 'std-10x', 'geneid-SS2', 'chromosome-SS2', 'is_mito-SS2', 'highly_variable', 'mean', 'std'
uns: 'DM', 'DM_EigenValues', 'anno_man_colors', 'biosample_id_colors', 'data_type_colors', 'density_predictor', 'diffmap_evals', 'fine_anno_colors', 'iroot', 'isroot_colors', 'leiden', 'leiden_DM_colors', 'leiden_DM_sizes', 'leiden_colors', 'leiden_sizes', 'neighbors', 'original_pop', 'paga', 'pagaDM', 'pagaPCA', 'pca', 'phase_colors', 'pop', 'predicted_doublets_colors', 'pseudo_bulk', 'pseudo_bulk_settings', 'rank_genes_groups', 'rs50', 'umap_init_pos', 'umap_init_pos_DM'
obsm: 'DM_EigenVectors', 'DM_scaled', 'Delta_DM', 'TIGON_u', 'X_diffmap', 'X_pca', 'X_pca_harmony', 'X_pca_scaled', 'X_umap', 'X_umap_2d', 'X_umap_3d', 'X_umap_DM_2d', 'X_umap_DM_3d', 'mellon_u', 'surrogate_u'
varm: 'PCs'
obsp: 'DM_Kernel', 'DM_Similarity', 'DM_connectivities', 'DM_distances', 'connectivities', 'distances'
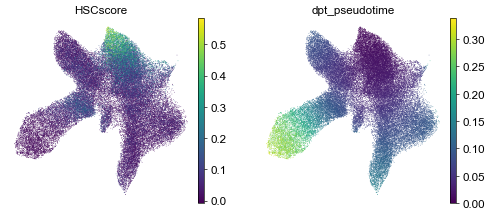
if 'X_diffmap' not in adata.obsm_keys():
# use batch corrected cell representation like Harmonied PC or SCVI latent
sc.pp.neighbors(adata, n_pcs=30, use_rep='X_pca_harmony')
sc.tl.diffmap(adata, n_comps=10)
adata.uns['iroot']= np.argmin(adata.obs['HSCscore'].values)
sc.tl.dpt(adata)
sc.pl.umap(adata, color=['HSCscore', 'dpt_pseudotime'])
# next we scaled the diffusion map to 0-1.
DM = adata.obsm['X_diffmap']
DM_min = DM.min(axis=0,keepdims=True)
DM_range = DM.max(axis=0,keepdims=True) - DM_min
DM_scaled = (DM - DM_min)/DM_range
Use palantir to compute DM
import palantir
dm_res = palantir.utils.run_diffusion_maps(adata, pca_key='X_pca_harmony')
ms_data = palantir.utils.determine_multiscale_space(adata)
print(f"shape of palantir DM : {adata.obsm['DM_EigenVectors'].shape} and scaled DM : {adata.obsm['DM_EigenVectors_multiscaled'].shape} ")
4. Sample local cell state changes in diffusion map¶
Pseudodynamics+ can make use of the local cell state changes in diffusion map to assists learning velocity, inspired by TrajectoryNet by Tong et. al.. This state changes is defined as \(\Delta x = \sum_{i \in N(x)} \frac{w_{i,x}}{w_{i,x} + \sum_{j \in N(x)} w_{i,j}} \Delta x_i\) ,Here we provdie two ways of sampling this local state transition:
by default we rely on KNN and pseudotime with \(w_{i,j}\) denoting the connectivities in KNN.
alternatively, when a transition matrix is provided, e.g. cellrank. The \(w_{i,j}\) denote the probability of transitioning from \(i\) to \(j\).
delta_DM , neighbors = pdp.tl.sample_deltax(adata, xkey='DM_scaled', pseudotimekey='dpt_pseudotime', repeat=10)
delta_DM_ay = np.stack(delta_DM)
print(delta_DM_ay.shape)
adata.obsm['Delta_DM'] = delta_DM_ay.mean(axis=-1) # use the averaged ∆DM
(49390, 10, 30)
5. train test split¶
def train_test_split_adata(adata, leaveout=None, val_size=0.1, test_size=0.1, timepoint_key='timepoint_tx_days'):
obs = adata.obs
val_test_cbs = obs.query(f"`{timepoint_key}` not in @leaveout").sample(frac=0.2).index
test_cb = np.random.choice(val_test_cbs, size=len(val_test_cbs)//2,replace=False)
split_mapper = {cb:'test' for cb in test_cb}
val_cbs = {cb:'val' for cb in val_test_cbs if cb not in test_cb}
train_cbs = {cb:'train' for cb in adata.obs_names if cb not in val_test_cbs}
split_mapper.update(val_cbs)
split_mapper.update(train_cbs)
return adata.obs.index.map(split_mapper)
adata.obs['split'] = train_test_split_adata(adata, leaveout=[3])
adata.write_h5ad("data/tom_pos.h5ad")
What’s next
Estimate single-cell density at observed time point (optional).
set up training configuration